Last Modified 17 December 1999
The ``Static'' Tight-Binding Program: Example XII
Finding the Electronic Density of States and Band Structure for Monatomic Cubic Crystals
Electronic structure calculations such as our tight-binding method determine the energy eigenvalues en at some point k in the first Brillouin zone. If we know the eigenvalues at all points k, then the band structure energy (the total energy in our tight-binding method) is just
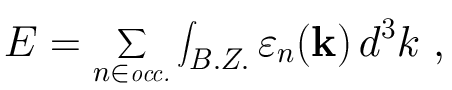
Alternatively, we might want to know the density of eigenvalues in the energy range [e,e+de], where de is small enough so that this density doesn't change much over this interval. This is known as the electronic density of states (DOS). It is obviously useful, since it tells you the energy states electrons can occupy in the crystal. In an insulator density of states actually vanishes at the Fermi level, meaning that electrons cannot move through the crystal without the application of a lot of energy.
If we know the band structure at every point in the Brillouin zone, then the DOS is given by the formula
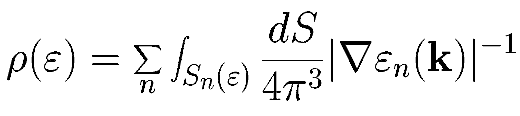
Obviously we can not evaluate this integral directly, since we don't know en(k) at all points, and can only guess at the properties of its gradient. One common approximation is to use the tetrahedron method, which divides the Brillouin zone into (surprise) tetrahedra, and then linearly interpolates within the tetrahedra to determine the gradient. This method is an approximation, but its accuracy obviously improves as we increase the number of k-points.
To begin this example, you will need the qlmtconvert.f and the dostet.f source files. The qlmtconvert converts the QLMT file created by the static program into a format that is usable by dostet. The dostet program will compute the Density of States (DOS) for the s, p, and d orbitals, along with the total DOS.
The dostet program takes as its input the dosdat.in file. The dosdat.in file is formatted as follows:
- The name of the element (e.g. "Ag", or "W", or "Re", etc.).
- The number of valence electrons and atomic weight of the element.
- The lattice constant in Bohrs.
- The type of k-point mesh to be used ( 2 or 4 for SC; 4 or 8 for BCC and FCC ), the number of bands to be computed, and the number of columns in the *.eng file (ALWAYS 6 in these examples).
- For SC, 2 → 35 k-points, 4 → 165 k-points
- For BCC, 4 → 55 k-points, 8 → 285 k-points
- For FCC, 4 → 89 k-points, 8 → 505 k-points
- The columns in the *.eng file correspond to the components of the Density-of-States: eigenvalue, s-state, p-state, d-state, 0, 0
- The minimum, step size, and maximum range of the DOS to be plotted.
An fcc crystal -- Silver
For our first example we will look at the electronic DOS and bandstructure of silver, a classic simple cubic material.
Create a directory fcc with the following files:
$ ls -al fcc/ total 108 -r--r--r-- 1 ssayed2 users 7130 Aug 17 13:42 ag_par -r--r--r-- 1 ssayed2 users 971 Aug 17 13:32 bandplot.in -rw-r--r-- 1 ssayed2 users 206 Aug 17 13:45 dosdat.in -rwxr-xr-x 1 ssayed2 users 3050 Aug 17 13:47 dosplot.ag.fcc.gnu -rw-r--r-- 1 ssayed2 users 25767 Aug 17 13:43 kpoints.fcc505 -rw-r--r-- 1 ssayed2 users 4195 Aug 17 13:43 kpoints.fcc89 -rw-r--r-- 1 ssayed2 users 1386 Aug 17 13:46 SKIN.band -rw-r--r-- 1 ssayed2 users 963 Aug 17 13:46 SKIN.dos -rw-r--r-- 1 ssayed2 users 10003 Aug 17 13:42 spcgrp.fcc
- Copy SKIN.dos as SKIN and run the static program. This example will use the kpoints.fcc89 file to read in the k-points. Later, you can try the kpoints.fcc505 file to see the difference in the output. If you use the kpoints.fcc505, then you need to modify line 4 of the dosdat.in file from 4 to 8.
- Now run qlmtconvert to convert the QLMT file to a different format, which will create the file dosapw.eng
- Finally, run the dostet to calculate the DOS. The dostet program will use the dosdat.in and dosapw.eng files as input.
- Use the gnuplot script provided to plot the results.
- Use ghostview to view the DOS plot.
$ cp SKIN.dos SKIN
$ static > output.dos
$ qlmtconvert
$ dostet
$ gnuplot dosplot.ag.fcc.gnu
$ gv dosdatall.eps
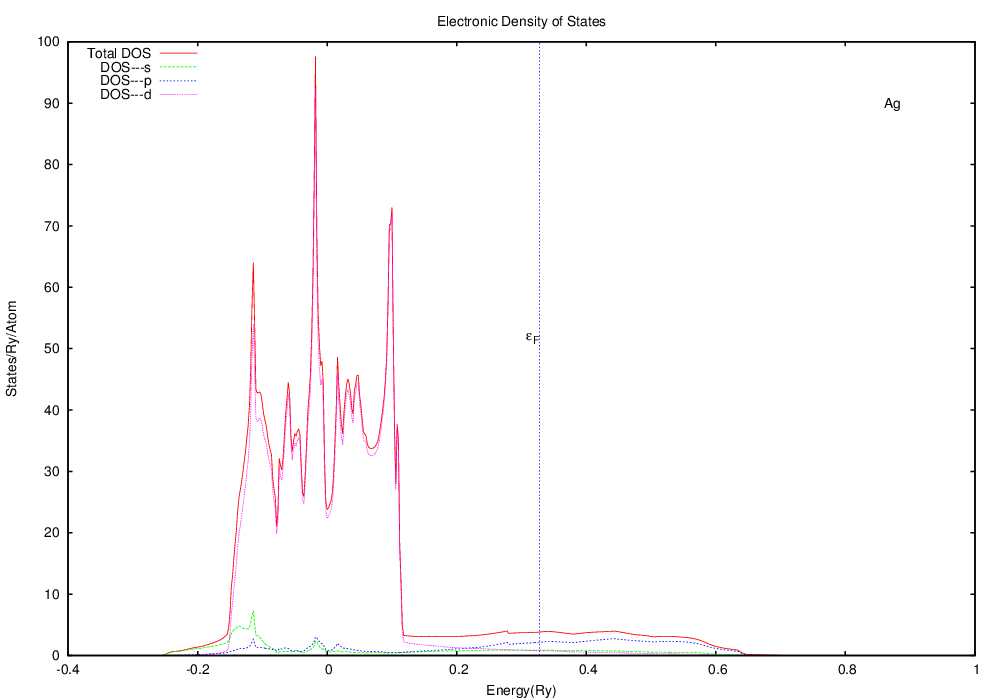
To create your own plots, I have provided a template gnuplot script that you can modify for your own purposes. At a minimum, you will need to find-replace the string "Elem" with the element symbol that you are working with (e.g. "Ag", "W", "Re", etc. ). You will also need to find-replace the string fermi with the calculated value of the Fermi energy. You can find the calculated value of the Fermi energy in the output file dosapw.itp, where it is listed as the first value in the file.
Now let's compute the bandstructure of Silver. For this, you will need the bandplot.f source, and bandplot.in.
- Copy SKIN.band to SKIN and run the static program to create a new QLMT file.
- Run the bandplot program to create bandplot.gnu. The bandplot program will create the gnuplot script for you.
- Use the generated gnuplot script to plot the bandstructure.
- Use ghostview to view the bandstructure plot.
$ cp SKIN.band SKIN
$ static > output.band
$ bandplot
$ gnuplot bandplot.gnu
$ gv bandplot.eps
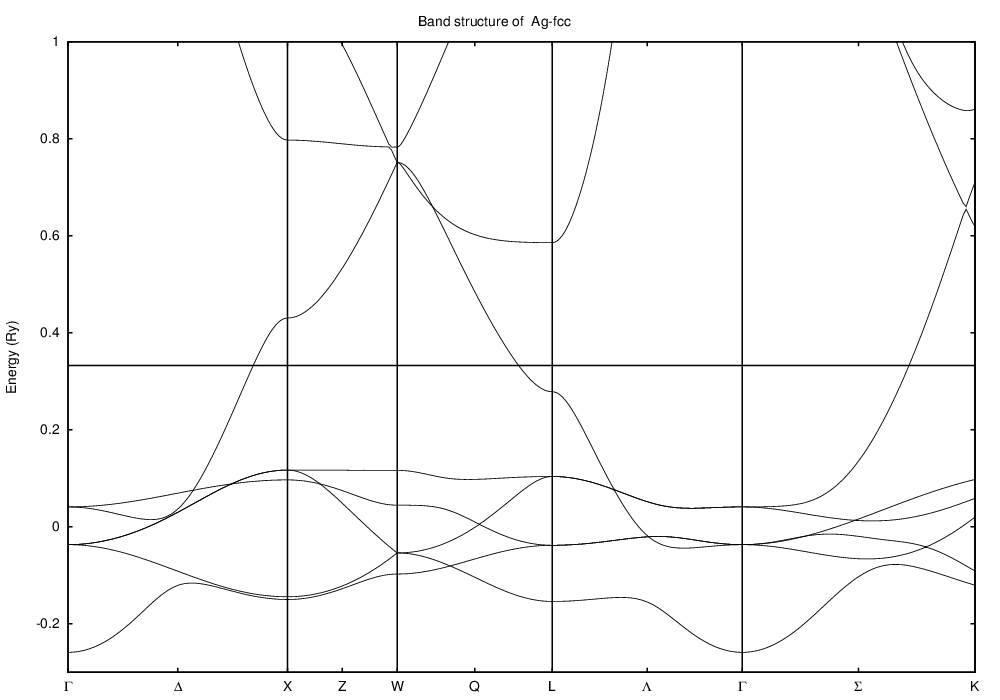
This last picture represents the band structure along certain
high-symmetry lines in the Brillouin zone of the fcc lattice:
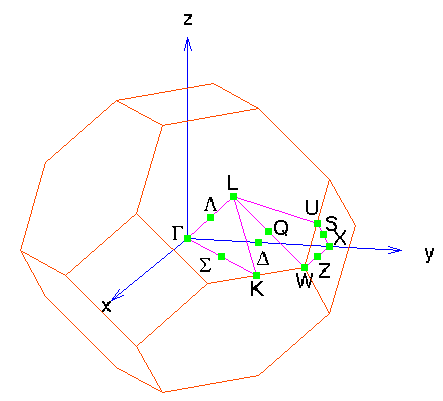
A bcc crystal -- Tungsten
That was relatively straight-forward. Now we want to do something similar for tungsten. This is a body-centered cubic metal with an equilibrium lattice constant of 3.16 Å. Get the parameter file w_par from the Tight-Binding Parameters page. I recommend doing this calculation in a new directory.
Create a directory bcc with the following files:
$ ls -al bcc/ total 208 -rw-r--r-- 1 ssayed2 users 1147 Aug 19 13:40 bandplot.in -rw-r--r-- 1 ssayed2 users 189 Aug 17 13:48 dosdat.in -rwxr-xr-x 1 ssayed2 users 3049 Aug 17 13:48 dosplot.w.bcc.gnu -rw-r--r-- 1 ssayed2 users 13996 Aug 17 13:47 kpts.bcc285 -rw-r--r-- 1 ssayed2 users 2541 Aug 17 13:47 kpts.bcc55 -rw-r--r-- 1 ssayed2 users 1391 Aug 17 13:49 SKIN.band -rw-r--r-- 1 ssayed2 users 962 Aug 17 13:49 SKIN.dos -rw-r--r-- 1 ssayed2 users 10003 Aug 17 13:48 spcgrp.bcc -r--r--r-- 1 ssayed2 users 7131 Aug 17 13:47 w_par
- Copy SKIN.dos as SKIN and run the static program.
- Now run qlmtconvert to convert the QLMT file to a different format.
- Finally, run the dostet to calculate the DOS.
- Use the gnuplot script provided to plot the results.
- Use ghostview to view the DOS plot.
$ cp SKIN.dos SKIN
$ static > output.dos
$ qlmtconvert
$ dostet
$ gnuplot dosplot.w.bcc.gnu
$ gv dosdatall.eps
The DOS should look something like this:
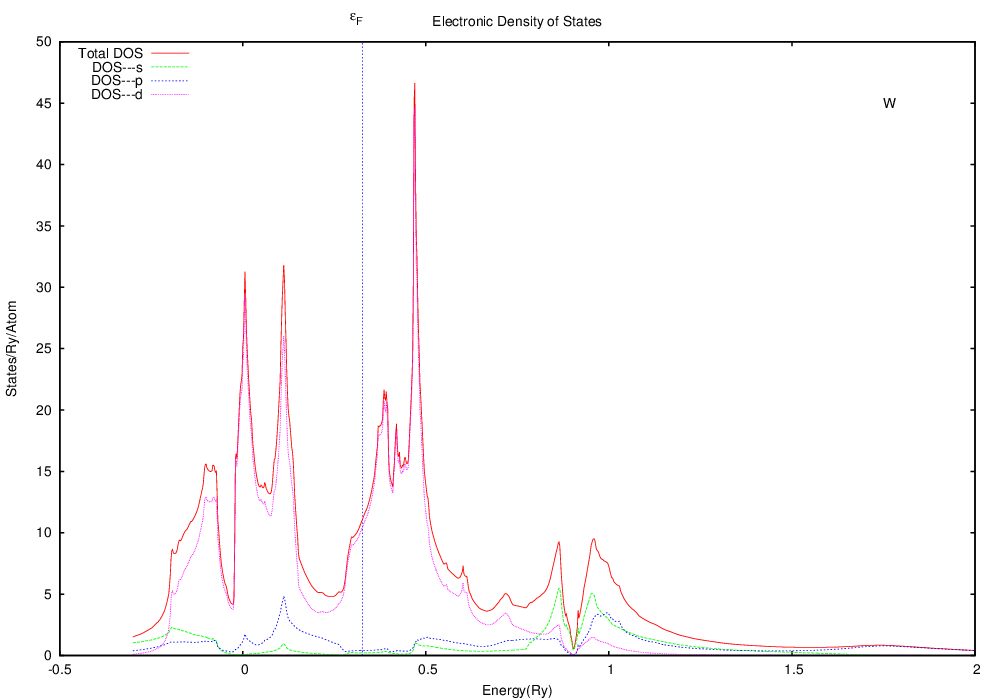
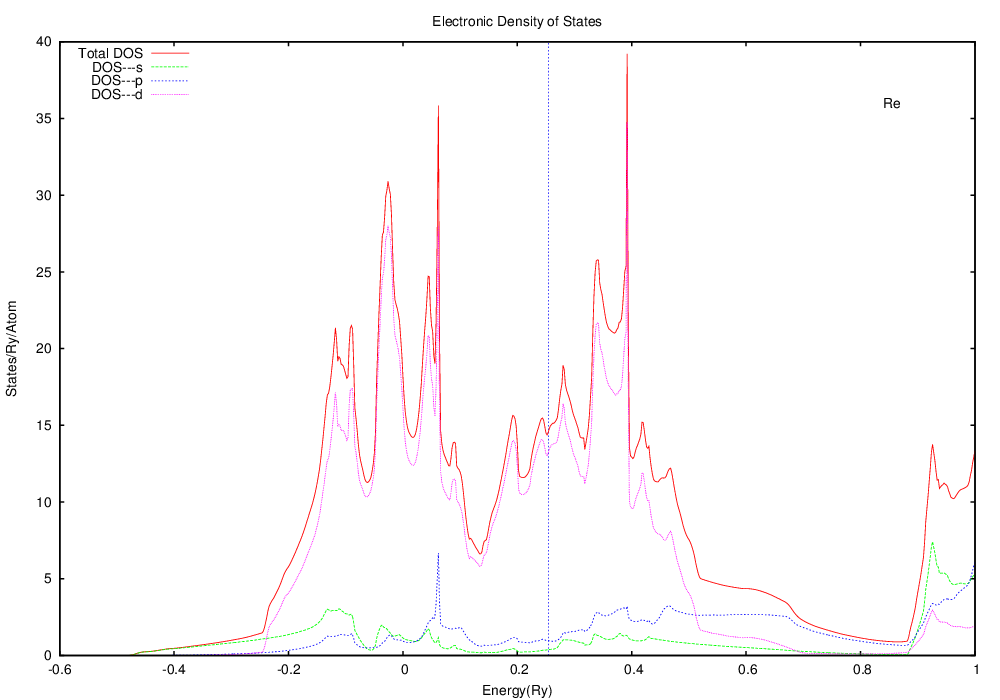
Note that this is substantially different from silver. The Fermi level (the highest state occupied by electrons) is well within the d bands, in contrast to silver, where the Fermi level was in a region with little contribution from the d states. If you think that this may have something to do with silver being a ductile fcc crystal and tungsten being a somewhat brittle bcc crystal, you're right.
Now let's compute the bandstructure of Tungsten. For this, you will need the bandplot.f source, and bandplot.in.
- Copy SKIN.band to SKIN and run the static program to create a new QLMT file.
- Run the bandplot program to create bandplot.gnu
- Use the generated gnuplot script to plot the bandstructure.
- Use ghostview to view the bandstructure plot.
$ cp SKIN.band SKIN
$ static > output.band
$ bandplot
$ gnuplot bandplot.gnu
$ gv bandplot.eps
The band structure for bcc materials is ploted along these
high-symmetry directions:
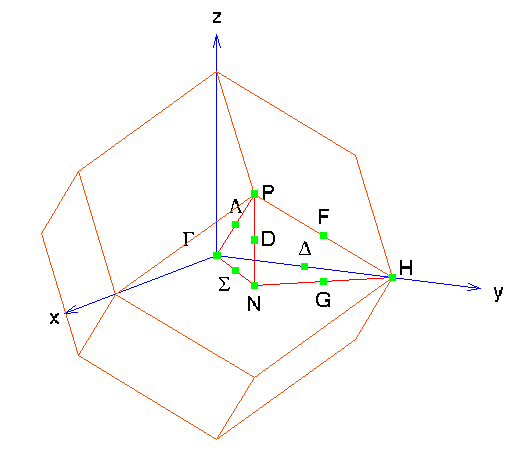
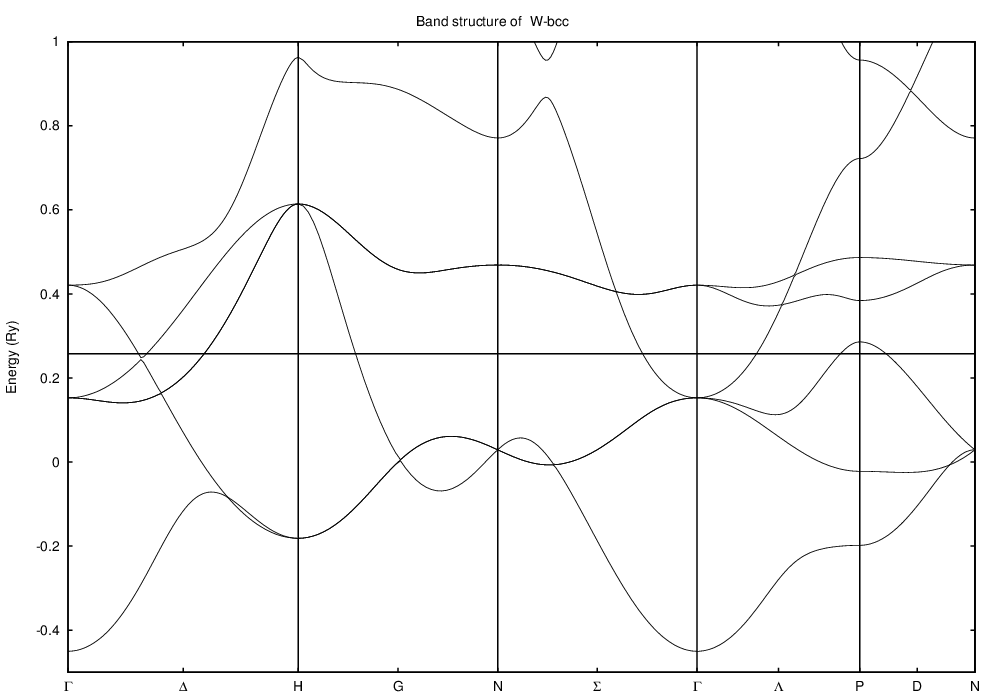
and looks like this for tungsten:
A hypothetical simple cubic crystal -- Rhenium
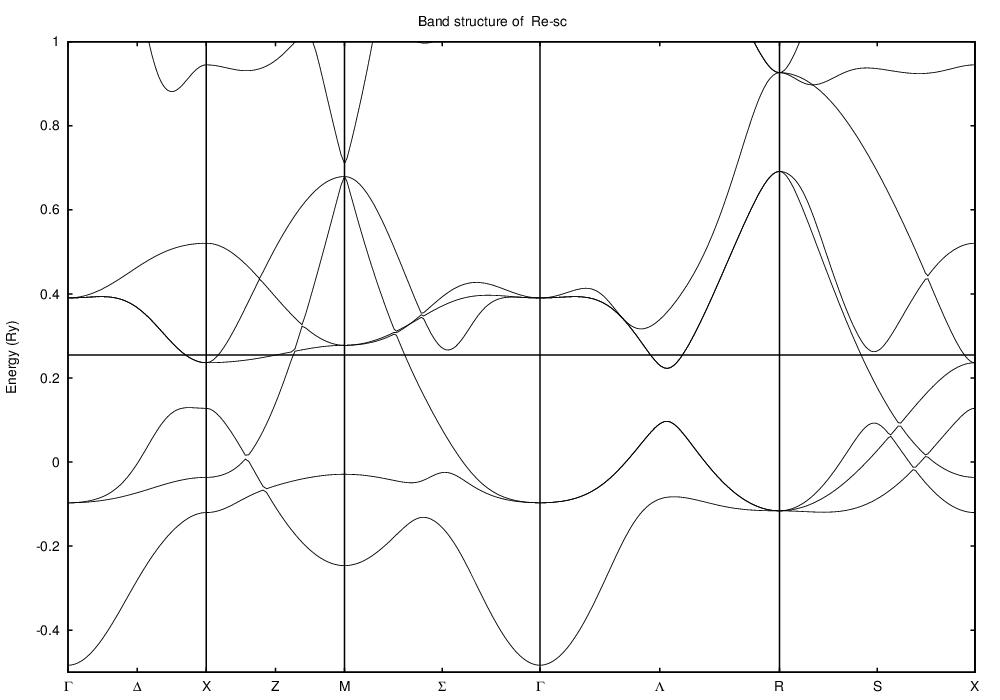
The only element which has a simple cubic ground state is Polonium, something we have not parameterized. So I'll take a hypothetical simple cubic structure of, say, Rhenium, with parameters re_par, and look at its DOS and band structure in a simple cubic lattice. To get a lattice constant, note that in its equilibrium hexagonal close-packed structure each Rhenium atom occupies 14.7 Å3. This corresponds to a simple cubic lattice constant of 2.45 Å. Since a simple cubic structure is usually less dense than a close packed structure, let's do the calculation at 2.60 Å.
For this example, I'll leave it to you to run the right program, and I will just show the results. The necessary files are listed below:
Create a directory sc with the following files:
$ ls -al sc/ total 160 -rw-r--r-- 1 ssayed2 users 1144 Aug 24 16:17 bandplot.in -rw-r--r-- 1 ssayed2 users 189 Aug 24 16:17 dosdat.in -rwxr-xr-x 1 ssayed2 users 3081 Aug 24 16:17 dosplot.re.sc.gnu -rw-r--r-- 1 ssayed2 users 9247 Aug 24 16:18 kpts.sc165 -r--r--r-- 1 ssayed2 users 7131 Aug 24 16:18 re_par -rw-r--r-- 1 ssayed2 users 1384 Aug 24 16:18 SKIN.band -rw-r--r-- 1 ssayed2 users 957 Aug 24 16:18 SKIN.dos -rw-r--r-- 1 ssayed2 users 10003 Aug 24 16:18 spcgrp.sc
The DOS looks like this:
This is a diagram showing the high-symmetry directions of the
simple cubic Brillouin zone:
and here is the band structure along those points:
Now you can do this for all the other elements.
Look at other examples.
Get other parameters from the Tight-binding periodic table.
Return to the static Reference Manual.
The appearance of external hyperlinks does not constitute endorsement by the United States Department of Defense, the United States Department of the Navy, and the Naval Research Laboratory of the linked web sites, or the information, products or services contained therein. For other than authorized activities such as military exchanges and Morale, Welfare, and Recreation (MWR) sites, the United States Department of Defense, the Department of the Navy, and the Naval Research Laboratory does not exercise any editorial control over the information you may find at these locations. Such links are provided consistent with the stated purpose of this DoD web site.
Return to CCMS home page