Finding minimum energy c/a at fixed volume
In a cubic material, such as Palladium, finding the equation of state is easy: just enter a volume Vi, calculate Ei=E(Vi), repeat for awhile, and use a Birch (or another) fit on the set {Vi,Ei} to determine the equilibrium point and the EOS. In a hexagonal material it isn't quite that simple, because we have to find the minimum of the energy function E(V,c/a) at fixed volume. That is, for every volume Vi, we have to find the value (c/a)i which minimizes the total energy of at that volume, and then construct Ei=E(Vi,(c/a)i). Then we can fit the {Vi,Ei} pairs. Fortunately, in an hcp lattice, the atoms always remain at lattice coordinates (1/3,2/3,1/4) and (2/3,1/3,3/4), so we don't have to minimize the energy with respect to atomic positions.
There are many ways to perform the minimization. One of the easiest ways is based on a quasi-Newtonian method based on a script I call qnewt , which I've saved as work/eos/qnewt. This script takes a volume and a guess at the equilibrium c/a ratio for that volume as its input. It then gives you a better guess at the equilibrium c/a ration for that volume. You can use this guess in a new run of qnewt, and keep iterating until you get the equilibrium to the desired accuracy. Typically you can get the equilibrium energy to within 10-5 Rydbergs in two or three steps.
To see how this works, let's pick an arbitrary volume, say 180 Bohr3, and run qnewt on that. We also need a guess at the equilibrium c/a ratio. The experimental value is 1.586, so we'll start with that.
$ qnewt 180 1.586 1.57014000 180.000000 .153494107 .035772783 1.58600000 180.000000 .153334993 .034924053 1.60186000 180.000000 .153130047 .034338014 180.00000 1.62931217 0.033944498
The first three lines of output are the contents of an SKENG file, showing the energy as a function of c/a for three different values: 1.57014 (=0.99*1.586), our initial guess of 1.586, and 1.60186 (=1.10*1.586). Looking at the energies, we see that the minimum energy c/a ratio is probably going to be closer to 1.6 than 1.586. In fact, qnewt fits these three points to a parabola, and then finds that the minimum of this parabola is at 1.62931217, and the minimum value is 0.033944498. All of this information is printed out on the next line. It is unlikely that this is the correct minimum for our system, but it is a good next guess, so we'll try it again:
$ qnewt 180 1.62931217 1.61301905 180.000000 .152952943 .034081198 1.62931217 180.000000 .152641401 .033929475 1.64560529 180.000000 .152271288 .034030406 180.00000 1.63094990 0.033928199
Now at least we've bounded the minimum, and the predicted minimum energy (0.033928199) is very near the minimum energy we've found so far (0.033929475). We could stop here, but one more iteration isn't a bad idea, just to make sure we're doing things correctly:
$ qnewt 180 1.63094990 1.61464040 180.000000 .152924767 .034054374 1.63094990 180.000000 .152606687 .033928436 1.64725940 180.000000 .152230851 .034054143 180.00000 1.63095738 0.033928436
Now the predicted minimum energy and the minimum energy we've found are equal (0.033928436), so we are probably as close to the minimum as we can get. Now, if you look in your work/eos directory, you'll find a file named skeng.180, which contains all of the results of the SKENG files you've created for this volume:
1.57014000 180.000000 .153494107 .035772783 1.58600000 180.000000 .153334993 .034924053 1.60186000 180.000000 .153130047 .034338014 1.61301905 180.000000 .152952943 .034081198 1.62931217 180.000000 .152641401 .033929475 1.64560529 180.000000 .152271288 .034030406 1.61464040 180.000000 .152924767 .034054374 1.63094990 180.000000 .152606687 .033928436 1.64725940 180.000000 .152230851 .034054143
Keep this file around, as we'll need it in a few minutes.
Finding Ei = E(Vi,(c/a)i)
We now want to find the ``energy'' as a function of volume for this hcp lattice. By ``energy'', we mean ``lowest possible energy at this volume,'' and we determine this by the above procedure. Only now we'll do it for several volumes. Rather than bore you with all the calculations, I'll just leave the corresponding skeng files here and let you look at them. You can also run these calculations yourself using the qnewt script and the prototype SKIN file, skin.1. Save these files in your work/eos directory and you are ready to run. (If you are impatient, use the script qnewt3, which runs three iterations at a time.)
After calculations at several volumes, I now have a collection of skeng files:
$ cd work/eos $ ls -l skeng.* -rw-r----- 1 mehl users 567 Jun 4 10:43 skeng.150 -rw-r----- 1 mehl users 567 Jun 4 10:19 skeng.160 -rw-r----- 1 mehl users 567 Jun 4 10:22 skeng.170 -rw-r----- 1 mehl users 567 Jun 4 10:07 skeng.180 -rw-r----- 1 mehl users 567 Jun 4 10:26 skeng.190 -rw-r----- 1 mehl users 567 Jun 4 10:28 skeng.200 -rw-r----- 1 mehl users 567 Jun 4 10:34 skeng.210 -rw-r----- 1 mehl users 567 Jun 4 10:36 skeng.220 -rw-r----- 1 mehl users 567 Jun 4 10:37 skeng.230 -rw-r----- 1 mehl users 567 Jun 4 10:38 skeng.238 -rw-r----- 1 mehl users 567 Jun 4 10:39 skeng.240 -rw-r----- 1 mehl users 567 Jun 4 10:40 skeng.250 -rw-r----- 1 mehl users 567 Jun 4 10:41 skeng.260
We know that the each skeng file contains the minimum energy found at the volume, so we can use the script work/eos/findmin to sort through each file, find the c/a ratio containing the lowest energy, and send this line to the file work/eos/hcp.eng:
$ cat hcp.eng 1.64497271 150.000000 .195912899 .187430852 1.64184610 160.000000 .177130438 .114704213 1.63673261 170.000000 .163199291 .066240831 1.63094990 180.000000 .152606687 .033928436 1.62572142 190.000000 .144411879 .012838414 1.62101427 200.000000 .137941785 -.000118739 1.61695142 210.000000 .132735107 -.006950248 1.61328739 220.000000 .128506077 -.009018008 1.60960698 230.000000 .125062415 -.007287369 1.60658379 238.000000 .122770055 -.003661979 1.60579146 240.000000 .122252758 -.002485065 1.60174402 250.000000 .119954645 .004811185 1.59771104 260.000000 .118069641 .014124605
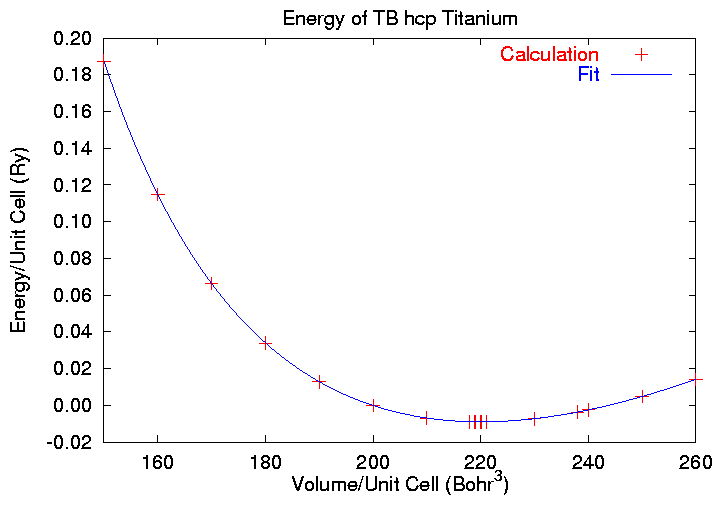
The minimum energy volume is near 220 Bohr3,
somewhat smaller than experiment but fairly consistent with
first-principles results. You'll note, though, that the equilibrium
c/a ratio is nowhere near the experimental value of 1.586. This is
an error in our tight-binding parametrization. We are considering
improvements in the parametrization which will improve the
calculated c/a value.
Now that we have a file containing
E(V), we can use a Birch fit to find the equilibrium energy and
bulk modulus. This is most easily done with a gnuplot script, work/eos/hcpfit.gnu, which
fits the total energy to a third-order Birch polynomial. Running
this script we get
Unfortunately, the predicted equilibrium energy is higher than
the known energy at V=220 Bohr3,
so we have to search around for the minimum a bit. Eventually we
come up with an equilibrium volume of 220.088 Bohr3,
and the file
work/eos/hcp.eng:
So, using our new equilibrium volume of
220.088 Bohr3 and equilibrium energy of
-0.009018141 Ry, we can do a higher-order Birch fit using the
script
work/eos/hcpfit2.gnu . This gives us the information:
and the plot:
So to summarize this section, we've found the following:
Go back to the Example X home page.
Look at other examples.
Get other parameters from the Tight-binding periodic
table. Return to the static Reference
Manual.
Finding the Equation of State and Equilibrium Properties
$ gnuplot hcpfit.gnu
[Fitting Information Deleted]
Equilibrium Volume = 218.825567968325 Bohr^3 = 32.426610660628 Angstroms^3
Equilibrium Energy = -0.00892653034413964 Ry
Equilibrium Bulk Modulus = 0.00821086196105331 Ry/Bohr^3 = 120.786019536211 GPa
dB/dP (P=0) = 5.26167550134072
Press Enter to end program
$
1.64497271 150.000000 .195912899 .187430852
1.64184610 160.000000 .177130438 .114704213
1.63673261 170.000000 .163199291 .066240831
1.63094990 180.000000 .152606687 .033928436
1.62572142 190.000000 .144411879 .012838414
1.62101427 200.000000 .137941785 -.000118739
1.61695142 210.000000 .132735107 -.006950248
1.61400746 218.000000 .129283772 -.008934964
1.61372104 218.800000 .128968824 -.008986750
1.61364943 219.000000 .128890893 -.008995800
1.61328739 220.000000 .128506077 -.009018008
1.61326030 220.088000 .128472502 -.009018141
1.61293422 221.000000 .128128916 -.009002417
1.60960698 230.000000 .125062415 -.007287369
1.60658379 238.000000 .122770055 -.003661979
1.60579146 240.000000 .122252758 -.002485065
1.60174402 250.000000 .119954645 .004811185
1.59771104 260.000000 .118069641 .014124605
$ gnuplot hcpfit2.gnu
Fit set for vo = 220.088 Bohr^3 and
Eo = -0.009018141 Ry. Change hcpfit2.gnu if necessary.
Press Enter to continue
[Fitting Information Deleted]
Equilibrium Volume = 220.088 Bohr^3 = 32.613683827428 Angstroms^3
Equilibrium Energy = -0.009018141 Ry
Equilibrium Bulk Modulus = 0.00827546824166049 Ry/Bohr^3 = 121.736411286626 GPa
dB/dP (P=0) = 3.80824655679457
Press Enter to end program
$
Experiment First-Principles
Tight-Binding a0 5.575
Bohr = 2.95 Å 5.424 Bohr = 2.87 Å
5.400 Bohr = 2.858 Å c0 8.844
Bohr = 4.68 Å 8.598 Bohr = 4.55 Å
8.715 Bohr = 4.611 Å V0 238.0
Bohr3 = 35.27 Å3
219.1 Bohr3 = 32.46 Å3 220.088 Bohr3 = 32.614
Å3 (c/a)0
1.586 1.585 1.613
B0 105 GPa
120 GPa 121.7 GPa