Last Modified 17 December 1999
In the fixed c/a calculation we looked at the energy as a function of the volume, V, at a fixed c/a ratio. In this section we'll turn that around and look at E(c/a) at fixed volume.
What do we get out of this? The answer lies in the theory of elasticity, to which we give short shrift here, and a bit longer explanation in a review article. Basically, if we work at constant volume, then changing the c/a ratio from the equilibrium value of (c/a)0 to c/a without changing the volume requires the strain parameters ei to obey the equations
| e2 | = | e1 | |
| (c/a)/(c/a)0 | = | (1+e3)/(1+e1) | (1) |
| (1+e1)2 (1+e3) | = | 1 |
If we define the relative change in c/a as
e1 = e2 = (1+z)-1/3 - 1 = -1/3 z + O(z2)
(3)
e3 = (1+z)2/3 - 1 = 2/3 z + O(z2)
Then as a function of z, the energy change can be related to the elastic constants by
E(z) = E0 + 1/9 V0 [ C11 + C12 + 2 C33 - 4 C13 ] z2 + O(z3) . (4)
Looking at our predefined strains table, we see that the situation described here is just strain type 4, except that we've written z instead of x. It's also the same strain we used in computing a Bain Path.
The path we want to take is then clear: we set up a SKIN file for an undistorted lattice starting with the ground state structure of our TB parameters for titanium, strain the crystal for several values of z (uh, x), and compute the elastic constant from
The SKIN file required for this job is stored in work/czz/SKIN . Since we still have not changed symmetry, it uses the same space group file (work/spcgp1.hcp), k-point mesh ( work/kmesh.194), and tight-binding parameters ( work/ti_par). The only thing new about the file is the response to the strain:
Mode=3 (Calculate energy only -- no pressure)
0.005 0.500 (T_{Fermi}, Eigenvalue cutoff for P calculation)
../ti_par
Titanium HCP (A3) -- Fixed Volume = 220.088 Bohr^3
1.61326030 -0.050 (We'll use the label to indicate c/a and strain)
0.00 (Electrons in addition to nominal Ti charge (=4/atom))
-3 (hexagonal lattice, read in V and c/a)
220.088 1.61326030 (V in Bohr^3 and c/a)
4 (c/a strain)
-0.050 (the strain factor)
2 (Atoms in the unit cell)
4 4 4 (Neighbor search cutoff indices)
F (Logical variable -- no internal displacements)
1 0.33333333333333333 0.66666666666666667 0.25 0 0 0 (Atom 1 in lattice coord.)
1 0.66666666666666667 0.33333333333333333 0.75 0 0 0 (Atom 2 in lattice coord.)
NEWSYM=T (Generate new set of k-points)
LATTIC=4 (Lattice type / Next is spacegroup file name:)
../spcgp1.hcp
ILAT=T (Space group file in lattice Coordinates)
-1313 (Read k-point generation information from file)
../kmesh.194
The strain type is ``4'', and we enter strain factors which range from -0.05 to +0.05. The final work/czz/SKENG file looks like this:
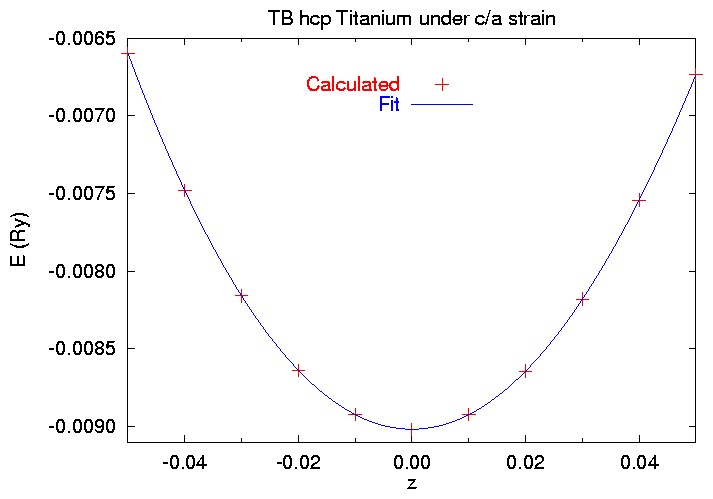
$ cat SKENG 1.61326030 -0.050 220.088000 .129360839 -.006598475 1.61326030 -0.040 220.088000 .129307485 -.007481490 1.61326030 -0.030 220.088000 .129184366 -.008159764 1.61326030 -0.020 220.088000 .128996025 -.008638850 1.61326030 -0.010 220.088000 .128754153 -.008923907 1.61326030 +0.000 220.088000 .128472502 -.009018141 1.61326030 +0.010 220.088000 .128162422 -.008923908 1.61326030 +0.020 220.088000 .127830874 -.008643724 1.61326030 +0.030 220.088000 .127479045 -.008181259 1.61326030 +0.040 220.088000 .127103646 -.007542400 1.61326030 +0.050 220.088000 .126700829 -.006735028
Since we know V0 and E0, and we know that z=0 is the equilibrium point, we can use gnuplot to fit this data to the polynomial
E(z) = E0 + 1/9 V0 z2 [ Czz + z ( A + z ( B + z C))] , (6)
where Czz is defined in (5) and A, B, and C are higher order elastic constants. When we run the fitting script we get
$ gnuplot !$ gnuplot czzfit.gnu Starting from v0 = 220.088 and e0 = -0.009018141 Change if appropriate Pressto continue [Fitting information deleted, see work/czz/fit.log] C_{zz} = 567.036295923323 GPa Press to quit $
and the fit looks like this:
So the result of this section is that we've found
Go back to the Example X home page.
Look at other examples.
Get other parameters from the Tight-binding periodic table.
Return to the static Reference Manual.