Last Modified 17 December 1999
So far we've looked at the energy of the hcp Titanium lattice as a function of
Is there anything else we can exploit, still keeping the full P63/mmc symmetry of the hcp lattice?It turns out that there is. In this section we'll take a look at the effects of changing the lattice constant a while leaving c fixed. In the next section we'll change c and keep a fixed.
As usual, the change in one of the lattice parameters of the hcp crystal is related to an elastic constant. If we change the lattice constant a, say by
a ---> a' = a (1 + x) , (1)
then according to our elastic constant tutorial we've constructed a strain
e1 = e2 = x ; e3-6 = 0 . (2)
This leads to the energy-strain relationship
E(x) = E0 + V0 (C11 + C12) x2 + O[x3] (3)
By now the drill should be pretty familiar: calculate E(x) at several points x, and determine the the elastic constant from a polynomial fit to the data. We'll do this in the directory work/c11+c12. The only tricky thing to note is that the pre-defined strain types table does not have an entry for the strain (2), so we'll have to use option ``1'' and provide all six of the ei. Thus the first calculation in the work/c11+c12/SKIN file looks like this:
Mode=3 (Calculate energy only -- no pressure)
0.005 0.500 (T_{Fermi}, Eigenvalue cutoff for P calculation)
../ti_par
Titanium HCP (A3) -- Fixed c
1.61326030 -0.050 (We'll use the label to indicate c/a and strain)
0.00 (Electrons in addition to nominal Ti charge (=4/atom))
-3 (hexagonal lattice, read in V and c/a)
220.088 1.61326030 (V in Bohr^3 and c/a)
1 (General strain)
-0.050 -0.050 0 0 0 0 (the strain factors)
2 (Atoms in the unit cell)
4 4 4 (Neighbor search cutoff indices)
F (Logical variable -- no internal displacements)
1 0.33333333333333333 0.66666666666666667 0.25 0 0 0 (Atom 1 in lattice coord.)
1 0.66666666666666667 0.33333333333333333 0.75 0 0 0 (Atom 2 in lattice coord.)
NEWSYM=T (Generate new set of k-points)
LATTIC=4 (Lattice type / Next is spacegroup file name:)
../spcgp1.hcp
ILAT=T (Space group file in lattice Coordinates)
-1313 (Read k-point generation information from file)
../kmesh.194
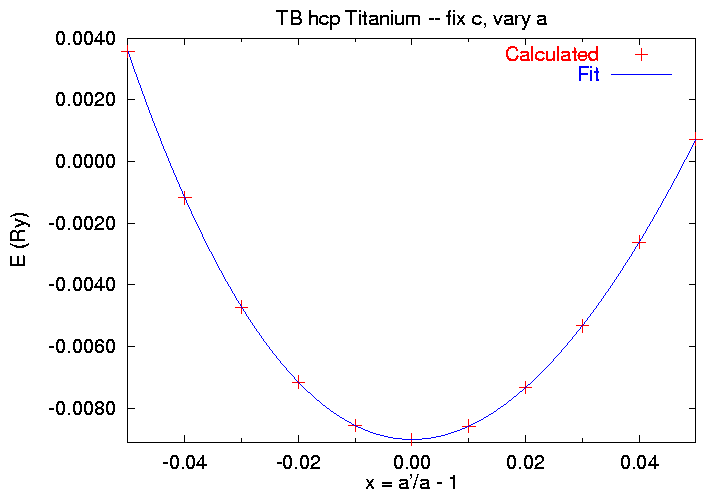
We use strain option 1, so we have to specify all six ei, according to (2). When we run the code for a full set of strains, we find
$ cat SKENG 1.61326030 -0.050 198.629420 .136781884 .003566595 1.61326030 -0.040 202.833101 .134873918 -.001177001 1.61326030 -0.030 207.080799 .133110223 -.004722904 1.61326030 -0.020 211.372515 .131464657 -.007158055 1.61326030 -0.010 215.708249 .129921976 -.008564840 1.61326030 0.000 220.088000 .128472502 -.009018141 1.61326030 0.010 224.511769 .127108770 -.008586483 1.61326030 0.020 228.979555 .125823896 -.007336256 1.61326030 0.030 233.491359 .124610983 -.005327283 1.61326030 0.040 238.047181 .123461329 -.002622256 1.61326030 0.050 242.647020 .122365654 .000721244
Note that it is much more difficult to compress the lattice than to expand it. We'll use a modification of the fitting procedure we used in the fixed volume case, using gnuplot to fit this data to the polynomial
E(x) = E0 + V0 x2 [ (C11 + C12) + x ( A + x ( B + x C))] , (4)
where A, B, and C are higher order elastic constants. When we run the fitting script we get
$ gnuplot caafit.gnu Starting from v0 = 220.088 and e0 = -0.009018141 Change if appropriate Pressto continue [Fitting information deleted. See fit.log] C_{11} + C_{12} = 295.490246436718 GPa Press to quit $
So in this section we've found that
Go back to the Example X home page.
Look at other examples.
Get other parameters from the Tight-binding periodic table.
Return to the static Reference Manual.