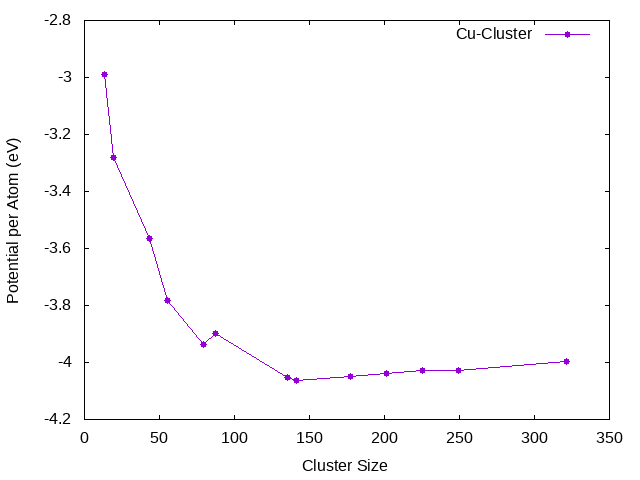
To demonstrate the calculation of cluster energies using the TBMD code, we use the example of the Cu system. The first step is to generate the cluster configurations. We can consider the fcc and bcc like clusters. The distances from a center atom in each of this clusters can be seen in Table 1. The atoms form shells around the central atom at these points. Once we have the atomic configurations of the clusters, we can set up the input files for the TBMD code.
| FCC | BCC | ||
| number | distance | number | distance |
| 1 | 0 | 1 | 0 |
| 12 | $\frac{1}{\sqrt{2}}$ | 8 | $\frac{3}{\sqrt{2}}$ |
| 6 | 1 | 6 | 1 |
| 24 | $\frac{\sqrt{6}}{2}$ | 12 | $\sqrt{2}$ |
| 12 | $\sqrt{2}$ | 24 | $\frac{\sqrt{11}}{2}$ |
| 24 | $\frac{\sqrt{10}}{2}$ | 8 | $\sqrt{3}$ |
| 8 | $\sqrt{3}$ | 6 | 2 |
| 48 | $\frac{\sqrt{14}}{2}$ | 24 | $\frac{\sqrt{19}}{2}$ |
| 6 | $2$ | 24 | $\sqrt{5}$ |
| 36 | $\frac{\sqrt{18}}{2}$ | 24 | $\sqrt{6}$ |
| 24 | $\sqrt{5}$ | 32 | $\frac{\sqrt{27}}{2}$ |
| 24 | $\frac{\sqrt{22}}{2}$ | 12 | $\sqrt{8}$ |
| 24 | $\sqrt{6}$ | 48 | $\frac{\sqrt{35}}{2}$ |
| 72 | $\frac{\sqrt{26}}{2}$ | 30 | $3$ |
With the cluster coordinates determined, we can now set up the TBMD input files. From the TBMD pages , we know that there are three input files needed,
in addition to the compiled tbmd code to run the calculation. The "INTBMD" file takes a slightly different form that what we would normally use in the TBMD simulations. This is shown below:system_name = "Cu fcc Cluster - conjugate gradient minimization" dt = 2.0 elec_gs_algo = 2 ionic_algo = 1 ! Conjugate gradient number_of_states = 7000 number_of_ionic_steps =100 calc_forces = .TRUE. sigma = 0.068 status_output_freq = 1 save_to_file = positions POS save_to_file = Energy ENE
Detailed description of the input files and general usage can be found on the TBMD pages. It is important to note here that the "ionic_algo" option is set to 1 in the INTBMD file. This changes the calculation to a Conjugate Gradient Relaxation calculation. The number of steps can be increased if the system does not converge within the set 100 steps.
The other file that needs to be set up is the "POSTBMD" file. We use the fcc-like cluster configuration as the crystal atomic positions. For a 13-atom cluster, the position file would look as below:
! 13 atom cluster of FCC Cu
a0 = 1.0
a_1 = 0.0000000 28.2400000 28.2400000
a_2 = 28.2400000 0.0000000 28.2400000
a_3 = 28.2400000 28.2400000 0.0000000
number_of_ions = 13
Positions = Cartesian
0.0000 0.0000 0.0000
0.0000 1.7650 1.7650
0.0000 -1.7650 1.7650
0.0000 1.7650 -1.7650
0.0000 -1.7650 -1.7650
1.7650 0.0000 1.7650
-1.7650 0.0000 1.7650
1.7650 0.0000 -1.7650
-1.7650 0.0000 -1.7650
1.7650 1.7650 0.0000
-1.7650 1.7650 0.0000
1.7650 -1.7650 0.0000
-1.7650 -1.7650 0.0000
The primitive cell is set to a large value so that the cluster atoms are in a large enough cell that isolates them from interacting with other atoms in a periodic system.
The codes for extracting bcc and fcc clusters for the noble metals and their outputs are listed below:
Cluster calculations have been done using the described method for Ag, Au, Cu, Pd and for CuAg.